Article 18, read-across from analogues with pre-2013 data, NAM-derived evidence anchored to defined developmental endpoints, and PBPK-informed IVIVE. When each is defensible to SCCS, and when it is not.
Article 18 of Regulation (EC) No 1223/2009 prohibits the marketing in the EU of cosmetic ingredients tested on animals for the purpose of meeting cosmetic safety requirements. The marketing ban for ingredient testing has been in force since 11 March 2009; the full marketing ban including repeated-dose, reproductive and toxicokinetic endpoints since 11 March 2013.
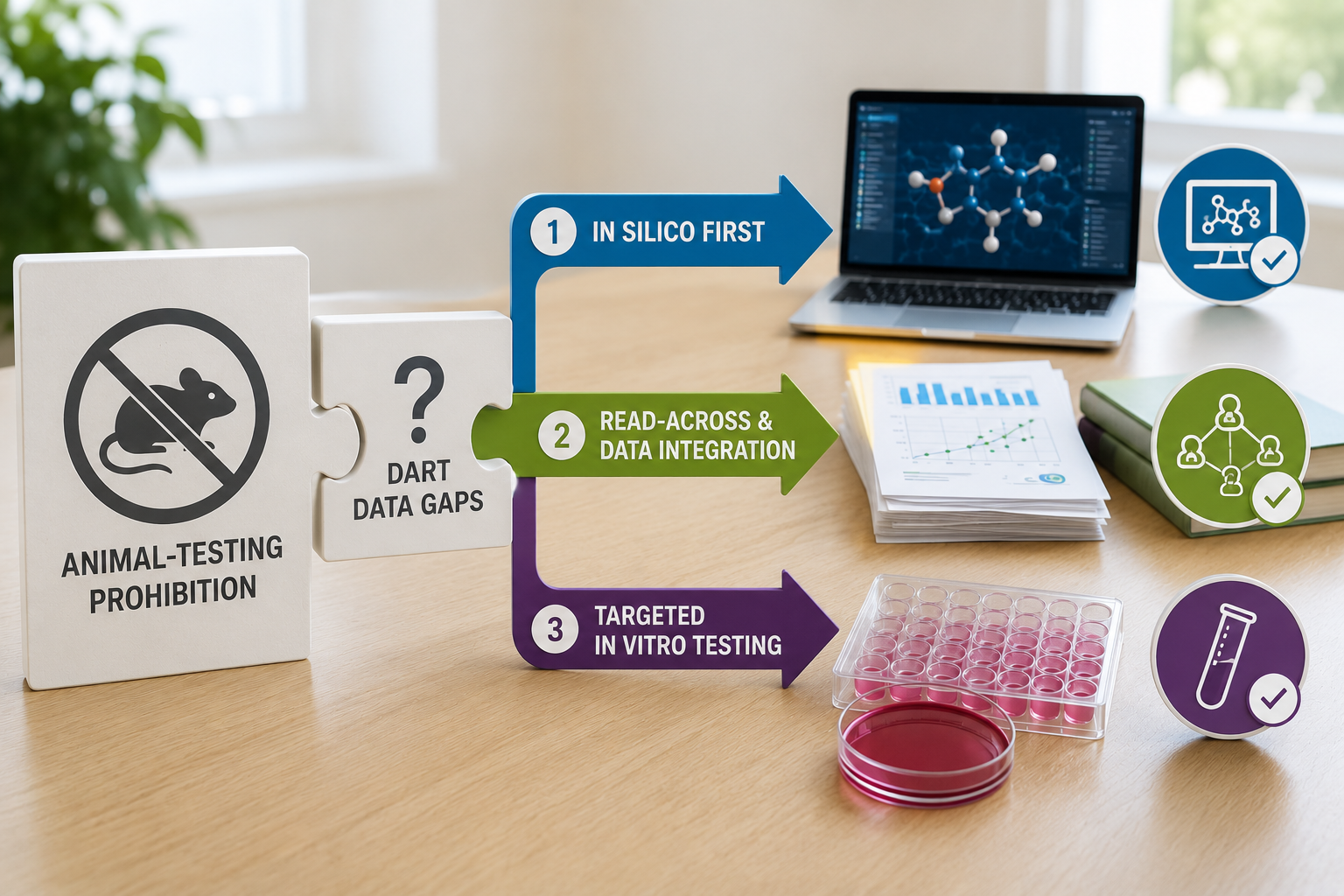
The challenge for cosmetic ingredients with limited toxicological data is that the standard battery for systemic safety assessment historically depended on developmental and reproductive toxicity (DART) studies in animals. Where these studies do not exist, and cannot be commissioned, an ingredient cannot be assessed by the conventional route. The 12th revision of the SCCS Notes of Guidance (SCCS/1647/22) prioritises animal-free new approach methodologies. The question is what those methodologies look like, in operational terms, when the endpoint is DART.
Three paths have emerged in practice. Each is defensible in some circumstances and not in others.
The most efficient path, where it is available, is read-across to one or more structurally and mechanistically related analogues with adequate pre-2013 DART data. The argument has to satisfy the structural similarity, toxicokinetic comparability, and metabolic relevance elements of a standard read-across justification, with particular attention to whether the placental transfer and developmental exposure profiles of the source and target are likely to be comparable.
The SCCS Notes of Guidance 12th revision is explicit that read-across is acceptable where supported by adequate similarity reasoning. The OECD Third Edition of the Guidance on Grouping of Chemicals (GD 418, December 2025) and EFSA's 2025 read-across guidance both reinforce the same principle: read-across is a hypothesis-driven scientific argument supported by empirical evidence on the source substance, mechanistic reasoning for transferability, and explicit uncertainty analysis.
Where this path fails, it usually fails for one of two reasons. The available analogues are too distant structurally, or their DART data quality does not support the conclusion the read-across is being asked to carry. In the first case, the read-across cannot be saved. In the second, the supporting dataset has to be strengthened through additional in vitro or in silico evidence on the analogue itself before it can support the target.
Where read-across is not available, in vitro and in silico methods can address specific developmental and reproductive endpoints directly. The available toolkit has expanded considerably in the past decade. Stem cell-based assays such as the embryonic stem cell test (EST) and ReproTracker (Toxys) assess perturbations in defined developmental pathways. The OECD-validated rat whole-embryo culture, zebrafish embryo toxicity assays, and high-throughput screening data from ToxCast and Tox21 covering reproductive and developmental targets all contribute to the available evidence base.
None of these methods, on its own, replaces a guideline-compliant DART study. Used in combination, anchored to a defined adverse outcome pathway, and integrated within a structured WoE framework, they can provide adequate evidence for an SCCS-aligned safety conclusion at realistic cosmetic exposure levels. Recent SCCS scientific opinions and industry case studies on cosmetic ingredients assessed through NAM-based approaches illustrate the structure of an acceptable DART argument under Article 18.
Where the toxicological mode of action is well-understood and the available NAMs cover the key events of the relevant adverse outcome pathway, the answer is sometimes yes. Where the mode of action is not characterised, or where the available NAMs do not address the developmental window of concern, NAM-only DART assessments remain difficult to defend.
The third path, which is increasingly used in combination with paths one and two, is quantitative in vitro to in vivo extrapolation (QIVIVE) supported by physiologically based pharmacokinetic (PBPK) modelling. The principle is straightforward. In vitro effect concentrations from NAM assays are translated to equivalent internal exposure levels in the human body, accounting for absorption, distribution, metabolism and excretion under realistic cosmetic use conditions. These internal effect concentrations are then compared with predicted internal exposure under intended product use, generating a margin of safety expressed in concentration terms rather than external dose.
PBPK-informed QIVIVE addresses two limitations of conventional NAM-based assessment. The first is the difficulty of relating in vitro effect concentrations to systemic exposure levels: the PBPK model translates between them. The second is the conservatism inherent in default cosmetic exposure assumptions: the model can be parameterised to reflect realistic skin penetration, hepatic first-pass effects, and elimination kinetics. The result is a quantitative comparison of pathway-perturbing concentration against expected internal concentration, supporting both an external (SED-based) and an internal (concentration-based) margin of safety.
This is the structure illustrated in the integrated NGRA case studies that have been presented at SCCS in recent years and discussed at workshops by Cosmetics Europe, Unilever and other industry groups. It is also the structure most likely to be defensible where the toxicological question requires translation between in vitro pathway perturbation and in vivo apical effects. The uncertainties (assay applicability domain, kinetic parameter assumptions, dose metric selection) have to be characterised explicitly, and the conclusion drawn against a margin of safety that accommodates them.
Three operational points follow from the experience of the past five years.
The methods are now mature enough to support that argument in many cases. The work is in deploying them with sufficient rigour to produce conclusions the SCCS, the Responsible Person, and the market-surveillance authority can stand behind.

Regulatory toxicologist with extensive cosmetic ingredient safety experience under Regulation 1223/2009. Recent publications on NAM-based assessment of prostaglandins and peptides for cosmetic use.